A 62-year-old African American female with PMHx of HTN, PUD and Graves’ disease s/ p radioactive iodine (RAI), presented to the ED for progression of bilateral vision loss to near-blindness over the past year. She began to lose her vision one year ago, a few months after receiving RAI. Her vision loss has been non-painful, gradual and associated with eye pruritis, irritation/foreign body sensation, lacrimation and occasional bursts of light.
The patient was seen in the eye clinic five months ago where she reported pain with eye movement. Her vision at that visit was 20/70 OS and 20/800 OS. Multifocal electrorentinography demonstrated her OD with severe central depression of waveform, consistent with scotoma and her OS with diffuse macular waveform depression. Orbital CT showed thickening of EOMs. She was seen two months later with a similar complaint; however, no ophthalmology consult was obtained because her symptoms were chronic. She followed up in eye clinic one and a half months later and was found to have profound progressive bilateral vision loss.
On ROS she denied any history of cancer, toxic exposures, HA, eye pain, or other neurologic symptoms. Medications included: synthroid, lisinopril, bupropion, amlodipine and atenolol. Social history was remarkable for being a remote smoker but no alcohol or drug abuse.
On physical examination, the patient was in no distress. Her vitals were BP 126/64, pulse 72, and temp 36.7 °C (oral) and respirations were 16 with SpO2 100%.
Her eyelids were proptotic with lid edema (photo below) and her conjunctiva were pink. On visual acuity exam, OD/OS, she was only able to visualize hand motion; unable to test visual fields or color vision; IOP OD 23/OS 25; pupils OD/OS 3–>2.5, brisk, no APD; EOMs were intact; one was able to see the lateral and medial rectus muscle insertion sites at end ROM (figures 2,3 on page 7).

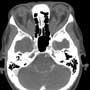
Ophthalmology was consulted and requested a CT head & orbits with contrast. CT showed diffuse enlargement of the extraocular muscles extending from the apex to the orbital attachment with mild enhancement in keeping with Grave’s ophthalmopathy (figures 3, 4). In addition, an incidental left petrous apex meningioma was demonstrated (figure 4).
These findings were consistent with compressive optic neuropathy and the patient was admitted for a 3-day course of IV Solumedrol 1g daily followed by po transition and taper. The patient underwent bilateral endoscopic orbital decompression by oculoplastic surgery due to acute worsening of Graves’ orbitopathy with orbital crowding. Two months postoperatively her pain had resolved and she was able to see color.

The physician was able to see the lateral and medial rectus muscle insertion sites at end ROM (left). CT showed diffuse enlargement of the extraocular muscles extending from the apex to the orbital attachment with mild enhancement in keeping with Grave’s ophthalmopathy (below).
Graves’ ophthalmopathy or orbitopathy is a syndrome of clinical and orbital imaging abnormalities with potentially sight-threatening ocular consequences. Although it generally occurs in patients with autoimmune hyperthyroidism, it is also known as dysthyroid ophthalmopathy or thyroid eye disease because occasionally it occurs in euthyroid, autoimmune hypothyroid disease states (Hashimoto’s thyroiditis) and thyroid dysfunction (due to Amiodarone). Rarely, it occurs in the absence of clinical or laboratory evidence of thyroid dysfunction and instead is associated with other autoimmune diseases (ex. Myasthenia gravis). Its incidence is approximately 16 women and 3 men per 100,000 population and it is the leading cause of proptosis in adults. Fifty percent of patients with Graves’ disease demonstrate some eye findings.

Graves’ disease affects orbital tissues, particularly the extra ocular muscles which are the primary focus of the disease. Of note, pupillary and ciliary muscles are always spared. Although eye complications usually manifest during hyperthyroidism, they may begin before there is any evidence of thyroid dysfunction or even after hyperthyroid treatment. Mucopolysaccharide deposition and chronic inflammatory cell infiltration in the EOMs cause the following: exophthalmos, lid retraction and lid lag, injection, conjunctival chemosis, episcleral inflammation, EOM weakness and subsequently in rare cases, diplopia, with poor eye movement, especially on upward gaze. These findings commonly are asymmetric and unilateral in appearance, at onset. To detect subtle exophthalmos, stand above and behind the seated patient and compare the relative positions of the lids and eyelashes. Ptosis in association with thyroid ophthalmopathy is usually due to coexistent myasthenia gravis, which may also contribute to ocular motility disturbance.

Concerning complications necessitating emergent treatment of Graves’ ophthalmopathy and ophthalmologic consultation include optic nerve compression and severe corneal exposure secondary to exophthalmos, both of which can lead to vision loss. Optic nerve compression at the orbital apex (secondary to EOM enlargement) may not present with concomitant significant exophthalmos. Early signs of optic nerve compression include afferent pupillary defect, impaired color vision and visual blurring that persists with eye closure, followed by loss of visual acuity and blindness if compression is unrelieved. Corneal exposure, leading to corneal ulceration or keratitis, results in periorbital edema and chemosis, eye pain, photophobia, conjunctival infection, visual loss and a flare of cells in the anterior chamber.
CT scan of the orbits commonly demonstrates enlarged extraocular muscles and less commonly, optic nerve distortion.
Maintaining a euthyroid state without an increase in TSH secretion is the primary treatment for ocular disease. If hyperthyroidism cannot be controlled by drug therapy, thyroid surgery is preferable, because Graves’ ophthalmopathy is more likely to worsen after radioiodine treatment. Cigarette smoking should be discouraged since this too increases the risk of orbitopathy progression. In mild to moderate disease, symptomatic treatment may also be implemented. Systemic steroids and/or other immunosuppresants are controversial. For corneal exposure, dark glasses and eye shields protect from light and dust. Elevating the head of the bed and using diuretics decrease periorbital and retro bulbar edema. Lubricants/artificial tears prevent corneal drying and topical adrenergic blockers (ex. guanethidine 5%) decrease lid retraction.
With optic nerve compression and corneal ulceration or keratitis, emergent treatment with high dose systemic steroids is required. Dosages of prednisone range from 1-2 mg/kg/d or 60-100 mg/d for 1 month, followed by a several month taper or IV pulse methylprednisolone, 1g/d for 3 days, repeated weekly for 3 weeks. In addition, “corneal ulcers,” with or without keratitis, require culture and topical antibiotics. If medical management is unsuccessful, either acutely or in the long term, it may be necessary to attempt retro bulbar irradiation, tarsorrhaphy (eyelids are partially sewn together to narrow the opening) for severe corneal exposure or orbit decompression (remove orbit roof) for optic nerve compression.